An overview of HIV-1 co-receptor function and its inhibitorsEmmanuel G. Cormier and Tatjana Dragic*
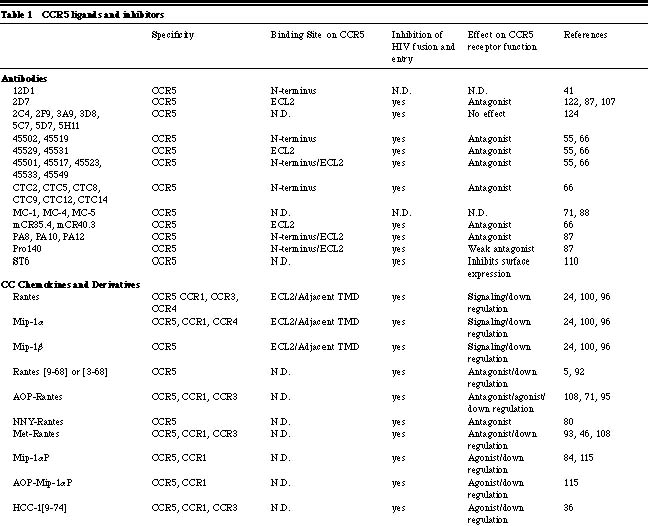
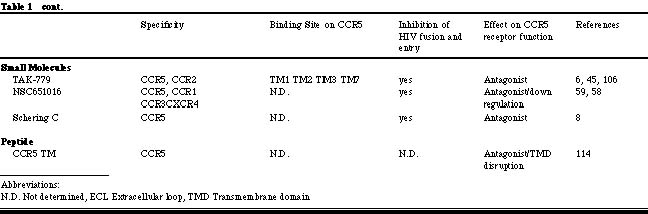
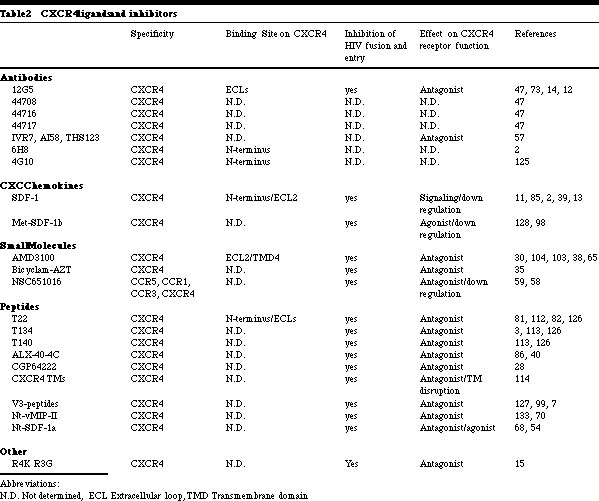
Albert Einstein College of Medicine, Department of Microbiology and Immunology 1300 Morris Park Avenue, Bronx, NY 10461 HIV-1 entry into target cells To trigger the membrane fusion process that leads to viral entry, HIV-1 must first interact with CD4 then with a co-receptor [1, 23, 42, 43, 51, 116, 121]. CD4 binding occurs subsequent to less specific, adhesion factor-mediated interactions with the cell surface that increase the localized concentration of virions [77]. Binding of the HIV-1 gp120 envelope glycoprotein to CD4 induces conformational changes in gp120 that create or expose a binding site for a co-receptor [116, 121]. Once available, the co-receptor binding site interacts with a complex, discontinuous region of the co-receptor that involves, but is not limited to the amino-terminal domain (Nt) [41, 44, 46, 91, 94, 97]. The association of gp120 with CCR5 or CXCR4 then drives additional conformational changes within the entire trimeric gp120/gp41 complex that eventually lead to the insertion of the gp41 fusion peptide into the host cell membrane, provoking fusion and entry [64]. HIV-1-co-receptor interactions HIV-1 co-receptors belong to the seven transmembrane G-protein coupled chemokine receptor family. The evidence accumulated to date indicates that there are similarities and differences in the way HIV-1 envelope glycoproteins from R5 and X4 isolates interact with their respective co-receptors. A cluster of residues in the CCR5 Nt participates in gp120-binding and is essential for fusion and entry of both R5 and R5X4 isolates [44, 49, 94]. In contrast, residues dispersed throughout the extracellular domain of CXCR4 are involved in gp120 docking, viral fusion and entry [18, 62]; each HIV-1 isolate uses a slightly different subset of CXCR4 residues in order to gain entry into the target cell. Nevertheless, the gp120 binding sites on CCR5 and CXCR4 comprise negatively charged and tyrosine residues [27, 44, 49, 50, 94]. Certain mutations in CXCR4 even enable it to mediate the entry of R5 isolates [16&endash;18, 120]. Similarities between CCR5 and CXCR4 gp120-binding sites are further underscored by the ability of R5X4 isolates to interact with both co-receptors. These similarities may account for the ability of a few residue changes in gp120 to induce a switch in co-receptor usage. It should be noted that the extracellular loops of CCR5 and CXCR4 also play an indirect role in viral entry by influencing the overall conformation and/or oligomerization of the co-receptor proteins. It is notable, however, that all chemokine receptors described to date have negatively charged regions in their extracellular domains, yet most do not mediate HIV-1 entry, and some do so only poorly. It also seems that the Nts of most if not all chemokine receptors contain sulfotyrosines. Hence, the unique features that make CCR5 and CXCR4 efficient HIV-1 co-receptors remain to be identified. Perhaps it is the way that the different Tyr-Asp-Glu motifs are exhibited on the surfaces of these receptors, or the ability of CCR5 and CXCR4 to interact with CD4 or other molecules on the cell surface that ultimately renders them efficient mediators of viral entry. Co-receptors and HIV-1 tropism The selective use of the CCR5 and/or CXCR4 co-receptors is the molecular explanation of the previous phenotypic categorization of HIV-1 isolates [9, 37, 52]. CCR5 is the principal co-receptor for HIV-1 variants that are sexually transmitted and persist within the majority of infected individuals (R5 isolates). The appearance of variants that use CXCR4 or both co-receptors (X4 and R5X4 isolates) signals accelerated CD4+ T-cell loss and disease progression [26, 109]. The phenotypic switch from R5 to X4 viruses in vivo typically occurs only after several years of infection. This is surprisingly slow given that changing only a few residues in gp120 can be sufficient to convert an R5 virus into an R5X4 virus in vitro and that such changes must be occurring continuously given the error rate of reverse transcription [19&endash;22, 25, 53, 60, 61, 64, 74, 78, 79, 90, 105, 116, 118, 119, 120]. These observations imply that the transition to CXCR4 usage is specifically suppressed in vivo [75]. The need for new inhibitors of HIV-1 replication Death rates due to HIV-1 infection have fallen significantly since specific inhibitors were developed that antagonize the function of the viral reverse transcriptase and protease enzymes [56]. Over a dozen different drugs now on the market target these two viral enzymes. However, the present antiviral cocktails are not well tolerated by a significant percentage (approximately 25%) of individuals [69]. There are also increasing concerns about the long-term metabolic side effects of protease inhibitors, notably poorly understand problems with fat metabolism [56, 69]. The increasing emergence and transmission of drug-resistant HIV-1 variants is another problem [129]. Together, the above factors emphasize the need to identify new classes of antiviral drugs that can supplement or partially replace existing drug cocktails. Among the many chemokine receptors that can mediate HIV-1 entry in vitro [10], only CCR5 and CXCR4 are of pharmacological importance, since they are the principal co-receptors used by HIV-1 to enter primary CD4+ T-cells and macrophages. These are the cells that produce almost the total viral burden in vivo [19, 76, 132]. In vitro experiments indicate that a lower level of CCR5 expression can reduce cellular infection by HIV-1, which may translate into clinical benefit [33, 123]. Blocking the function of CCR5 may not significantly impact human health since approximately 1% of Caucasians naturally lack CCR5 due to a protein-disrupting mutation without any detectable consequences [67, 101]. CCR5 does play a role in the correct functioning of the mammalian immune system, demonstrated by studies in CCR5 knock-out mice [134]. These animals have a greatly reduced survival after experimental infection of the brain with Cyptoccocus neoformans; partial defects in the clearance of Listeria monocytogenes; reduced IFN-g production after infection with Leishmania donovani; and an increased susceptibility to Toxoplasma gondii, due to decreased production of IL 12 and IFN-g. Whether this matters from the perspective of inhibitor development is uncertain. The safety of CXCR4 inhibitors may be more problematic in humans, because knocking out CXCR4 in mice is lethal [134]. However, a CXCR4-specific inhibitor was not acutely toxic in adult mice [29]. Co-receptor-targeted inhibitors of HIV-1 entry We have summarized the properties of the co-receptor inhibitors described in the literature to date in Tables 1 and 2. The first inhibitors known to prevent HIV-1 fusion and entry were MIP-1a, MIP-1b and RANTES, the natural CC-chemokine ligands of CCR5 [24]. The CXC-chemokine SDF-1a has an analogous inhibitory effect on viral entry via CXCR4 [11, 85]. Variants of chemokines with increased potency in vitro, usually resulting from N-terminal modifications to the RANTES or SDF-1a structure, have since been developed [5, 32, 68, 108, 128, 130]. Chemokines interfere with HIV-1 replication by several mechanisms: (1) direct competition between the chemokine and the gp120 glycoprotein for binding to the co-receptor, (2) a sustained down-regulation of the co-receptor as a consequence of chemokine binding and signal transduction, and (3) alteration of the differentiation state of the target cell that affects HIV-1 replication late in the viral life cycle [2, 4, 71, 89, 116, 117, 121]. Several CXCR4- and CCR5-specific murine MAbs are known to inhibit HIV-1 fusion and entry with considerable potency [47, 55, 87, 122]. Co-receptor specific MAbs are not agonists, but most are antagonists [47, 55, 87, 122]. We and others have shown that anti-CCR5 MAbs that recognize epitopes in the second extracellular loop (ECL2) are potent inhibitors of HIV-1 entry even though they only moderately inhibit gp120 binding to CCR5 [66, 87, 122]. Possibly, these MAbs inhibit important post-gp120 binding steps, such as conformational changes in CCR5 or its oligomerization [63]. Few anti-CXCR4 MAbs have been generated and only one has been extensively characterized. MAb 12G5 recognizes an epitope in ECL2 and inhibits HIV-1 fusion and entry both in an isolate- and a cell type-specific manner [73, 111]. Differences in gp120 affinities for CXCR4 and post-translational modifications of CXCR4 in different cell types could account for these discrepancies. Other anti-CXCR4 MAbs, whose epitopes remain to be determined, also variably inhibit the entry of the HIV-1 NL-43 isolate [57]. Several small molecule inhibitors of CXCR4- and CCR5-mediated HIV-1 entry are now known. All are receptor antagonists that have no signaling capacity themselves. The CXCR4 inhibitors T22, ALX40-4C and AMD3100 (and their derivatives) are highly cationic compounds, having at least six positively charged atoms at physiological pH [3, 38, 40, 81, 104]. The one small molecule CCR5 inhibitor whose structure has been described in print, TAK-779, has only one positive charge [6]. The difference in ligands probably reflects the surface charges of the two co-receptors; the CXCR4 surface is strongly anionic, whereas CCR5 has an almost neutral surface. T22, ALX40-4C and AMD3100 all bind predominantly to the extracellular domain of CXCR4, especially to the second extracellular loop, with anionic residues being of particular importance [65, 83]. All of these compounds antagonize signaling via SDF-1a [38, 40, 81, 104]. Surprisingly, each is completely specific for CXCR4 among other tested HIV-1 co-receptors, perhaps because of the unique nature of the CXCR4 extracellular surface. The binding site for TAK-779 on CCR5 is very different. Our mutagenesis studies have shown that TAK-779 binds within a pocket formed by transmembrane helices 1, 2, 3 and 7 of CCR5. Some contacts may also be made between TAK-779 and unidentified residues in the extracellular region, although this remains to be demonstrated. Once in place, TAK-779 prevents gp120, but not anti-CCR5 MAbs, from binding to CCR5 [6, 45]. TAK-779 is not an agonist, and does not cause CCR5 down-regulation but it does inhibit chemokine-induced receptor signaling [6, 45]. TAK-779 also interacts with CCR2b to block signaling and SIVrcm entry via this co-receptor [6, 45, 131]. The binding pocket for TAK-779 is probably not unique to CCR5 and similar pockets, likely to be present in other chemokine receptors, are attractive targets for drug development. HIV-1 escape from co-receptor-targeted inhibitors The following mechanisms of escape from co-receptor inhibitors are possible: (1) the escape mutant may continue to use the same co-receptor in an inhibitor-insensitive manner; (2) co-receptor switching may occur, so that an R5 virus now becomes able to use CXCR4, or vice versa; and (3) an entirely different co-receptor may now be used by the escape mutant. In the studies published to date, the first mechanism is the most common, while the third has not been found. The initial study on AMD3100 escape was performed before it was known that this compound targets CXCR4. It was carried out using HIV-1NL4-3 in MT-2 cells, which express CD4 and CXCR4, but not CCR5. The co-receptor-switching pathway was, therefore, unavailable. After 63 passages with increasing AMD3100 doses, the mutant virus had 13 amino acid changes scattered throughout gp120 and still used CXCR4, but in an AMD3100-insensitive manner [48, 102]. These observations imply that there may be more than one way for gp120 to functionally interact with CXCR4. A study with SDF-1a had a similar outcome: multiple amino acid changes in gp120 occurred during sequential passages to eventually create a resistant virus that still used CXCR4 for entry [102]. Interestingly, there was partial cross-resistance between the AMD3100- and SDF-1a resistant viruses, and about half of the amino acid substitutions in gp120 were common to the two escape mutants [102]. When AMD3100 resistance was selected for by using uncloned X4 or R5X4 primary isolates in PBMC (which express both CCR5 and CXCR4), R5 viruses become dominant in the cultures [48]. Escape mutant studies with CCR5-specific inhibitors have been more limited. An initial report using RANTES derivatives in hu-PBL SCID mice concluded that co-receptor switching to CXCR4 usage sometimes occurred [80]. However, escape from a CCR5-specific MAb in the same animal model did not involve co-receptor-switching (Moore, J.P. personal communication). Another study using MIP-1a and the R5 virus HIV-1JR-FL in a CCR5+ CXCR4+ cell line found that a 4-6-fold reduction in sensitivity to CC chemokines occurred after 3 months without any switch to CXCR4, despite its availability on the target cells [72]. Concluding remarksThe discovery of the principal HIV-1 co-receptors, CXCR4 and CCR5, has significantly impacted our understanding of how HIV-1 infects its target cells and how this relates to viral pathogenesis. New targets for antiviral drug development have been identified and are now being exploited. A CCR5 antagonist that blocked HIV-1 entry yet drove phenotypic evolution to CXCR4 use would be undesirable, because of the association between X4 and R5X4 viruses with an increased rate of CD4+ T-cell loss [26, 109]. However, a selection pressure seems to limit the rate of phenotypic evolution from CCR5 to CXCR4 use in vivo [75]. Furthermore, CCR5-negative individuals who became HIV-1 infected harbor exclusively CXCR4-using isolates [76]. The inability of these viruses to use CCR2b, CCR3, Bonzo, BOB, etc. indicates the irrelevance of these proteins in vivo. Unless a CCR5-specific inhibitor in some way interfered with the natural selection pressure that suppresses X4 viruses, blocking HIV-1 entry via CCR5 would not necessarily drive the rapid emergence of X4 viruses nor viruses that utilize other co-receptors [75]. CCR5 in particular and the co-receptors in general therefore represent viable drug targets aimed at slowing HIV-1 replication and the progression to AIDS.
|